1000 Genomes vs TOPMed Imputation in Samples of Diverse Ancestry
The hidden downside of better imputation results
The Stein Lab works with a unique set of human primary cell lines and donated tissue samples from a large set of donors. Each of these donors have been genotyped using standard genotyping platforms yielding over 1.7 million single nucleotide polymorphisms (SNPs). Overlapping these unique samples with individual genotypes from the HapMap3 project shows the diverse ancestry of this dataset.
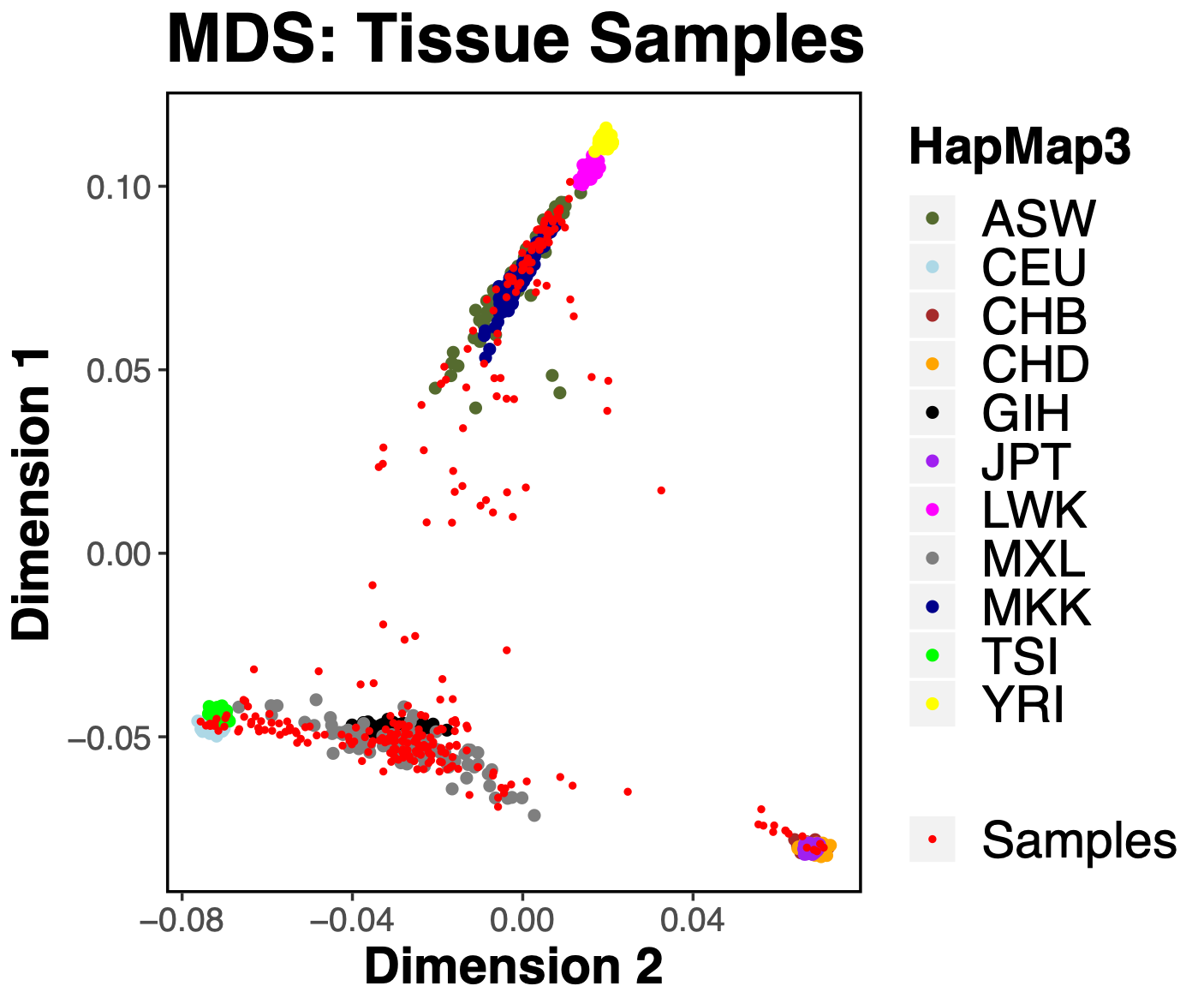
Before doing a genetic association analysis, it is common to increase the number of SNPs one wishes to test by first imputing the genotypes that are not measured by the genotyping arrays. In order to do this, we need a reference panel of genomes (or haplotypes) from a large set of individuals with similar ancestry to the samples we wish to impute. Since our samples are from a diverse ancestry, we need a reference panel which contains many representative populations. Two commonly used reference panels are the 1000 Genomes panel and the NHLBI Trans-Omics for Precision Medicine (TOPMed) panel.
- 1000 Genomes Phase 3 Version 5
- 5,008 haplotypes
- Diverse ancestry
- TOPMed Freeze 5
- 125,568 haplotypes
- Diverse ancestry
Both the 1000 Genomes and TOPMed reference panels contain individuals from a diverse ancestry. However, because the TOPMed reference panel contains significantly more individuals, we expect TOPMed to better impute diverse ancestry genotypes.
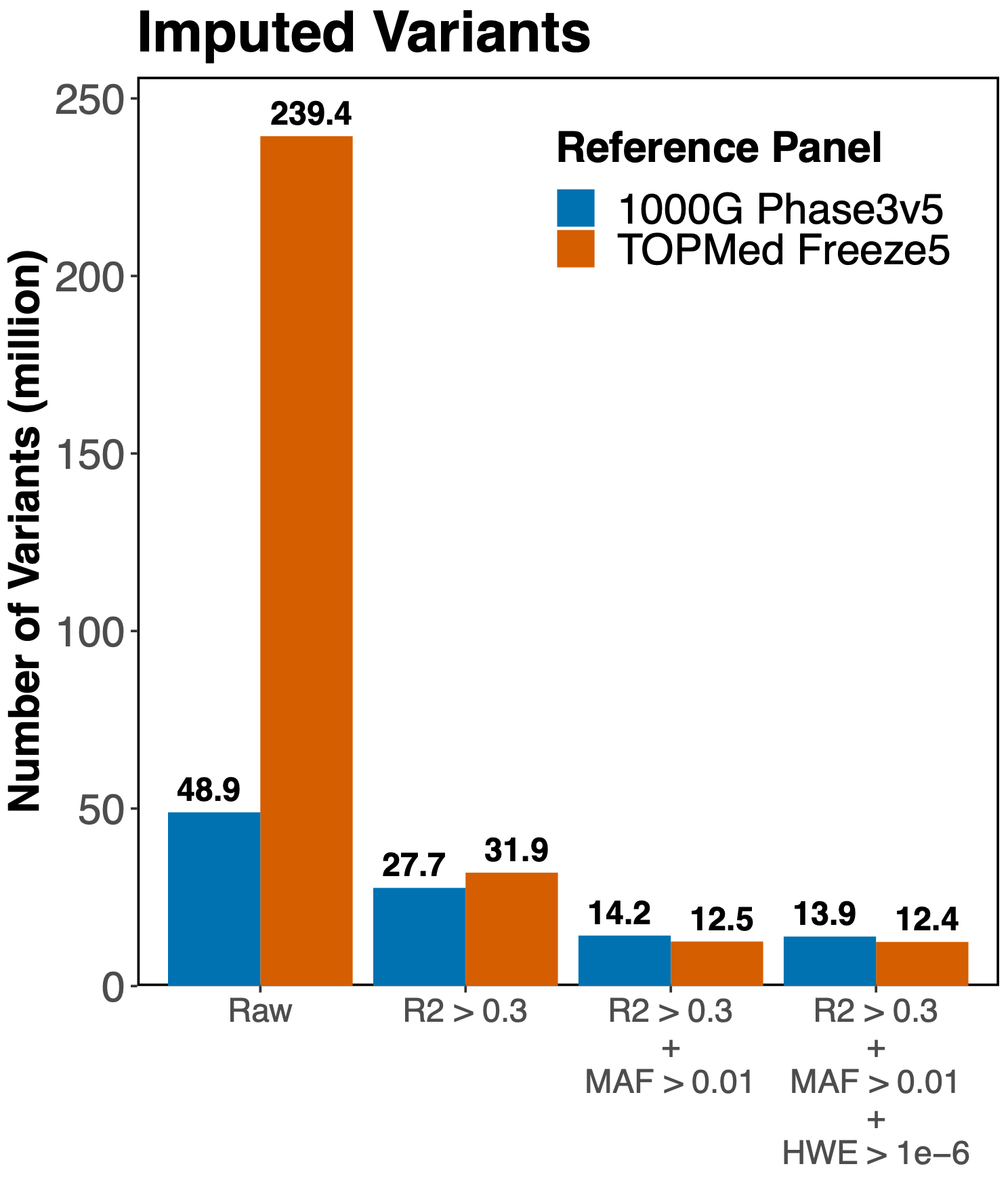
In this analysis, we used the Michigan Imputation Server to impute the genotypes of 326 diverse ancestry samples by starting with 1.7 million genotyped SNPs. At each imputed variant, the imputation quality was assessed with the r2 statistic (R2 or Rsq in the following graphs). An r2>0.3 was considered sufficient for use in downstream association analyses. At first glance, TOPMed yielded more high quality imputed variants than 1000 Genomes:
- 1000 Genomes
- 49 million imputed variants
- 28 million with an r2>0.3
- TOPMed
- 239 million imputed variants
- 32 million with an r2>0.3
Furthermore, when we look at variants across a range of minor allele frequencies (MAF), we see that TOPMed consistently yields better imputation quality than 1000 Genomes, more so for variants with an r2>0.3.
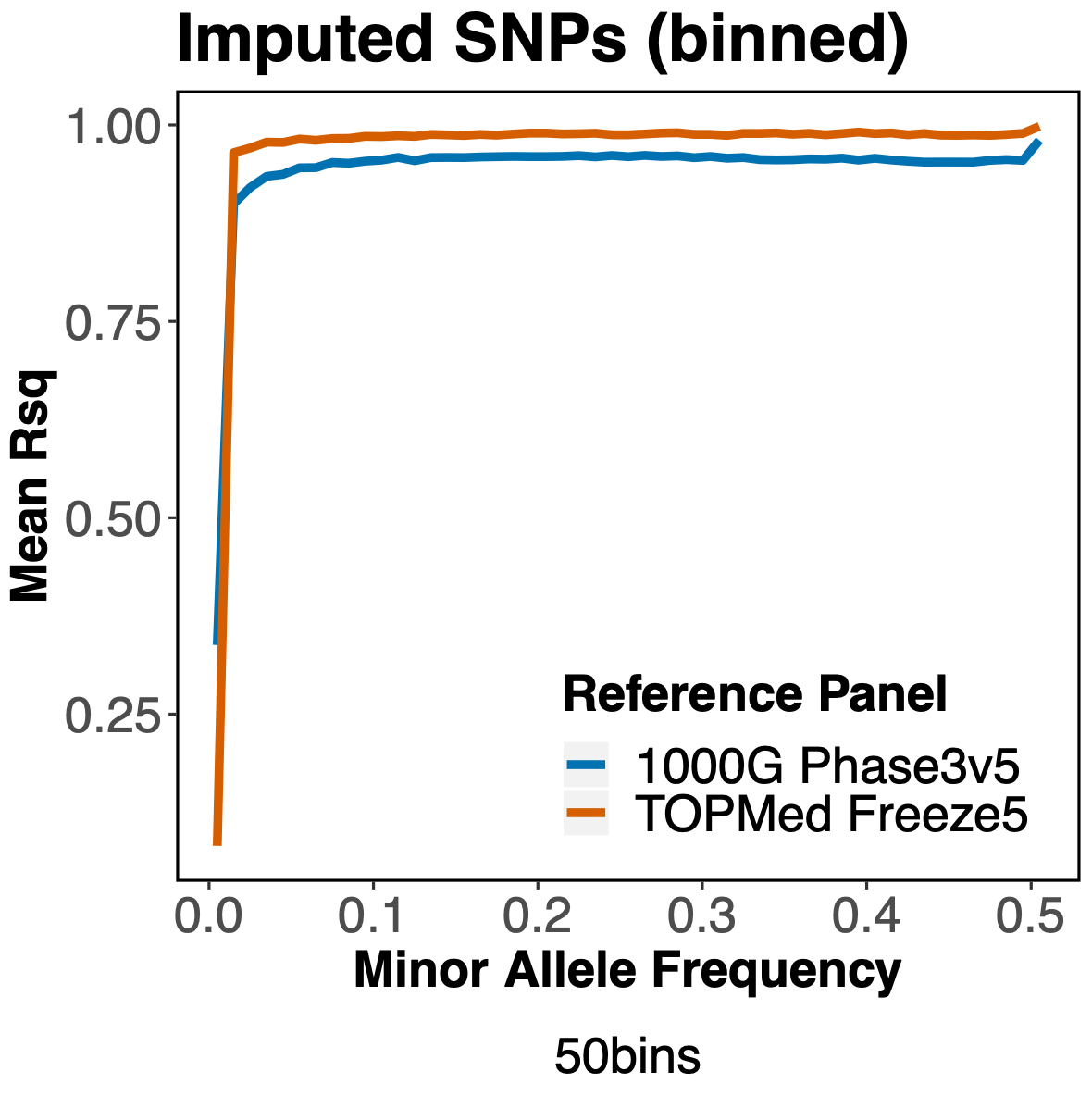
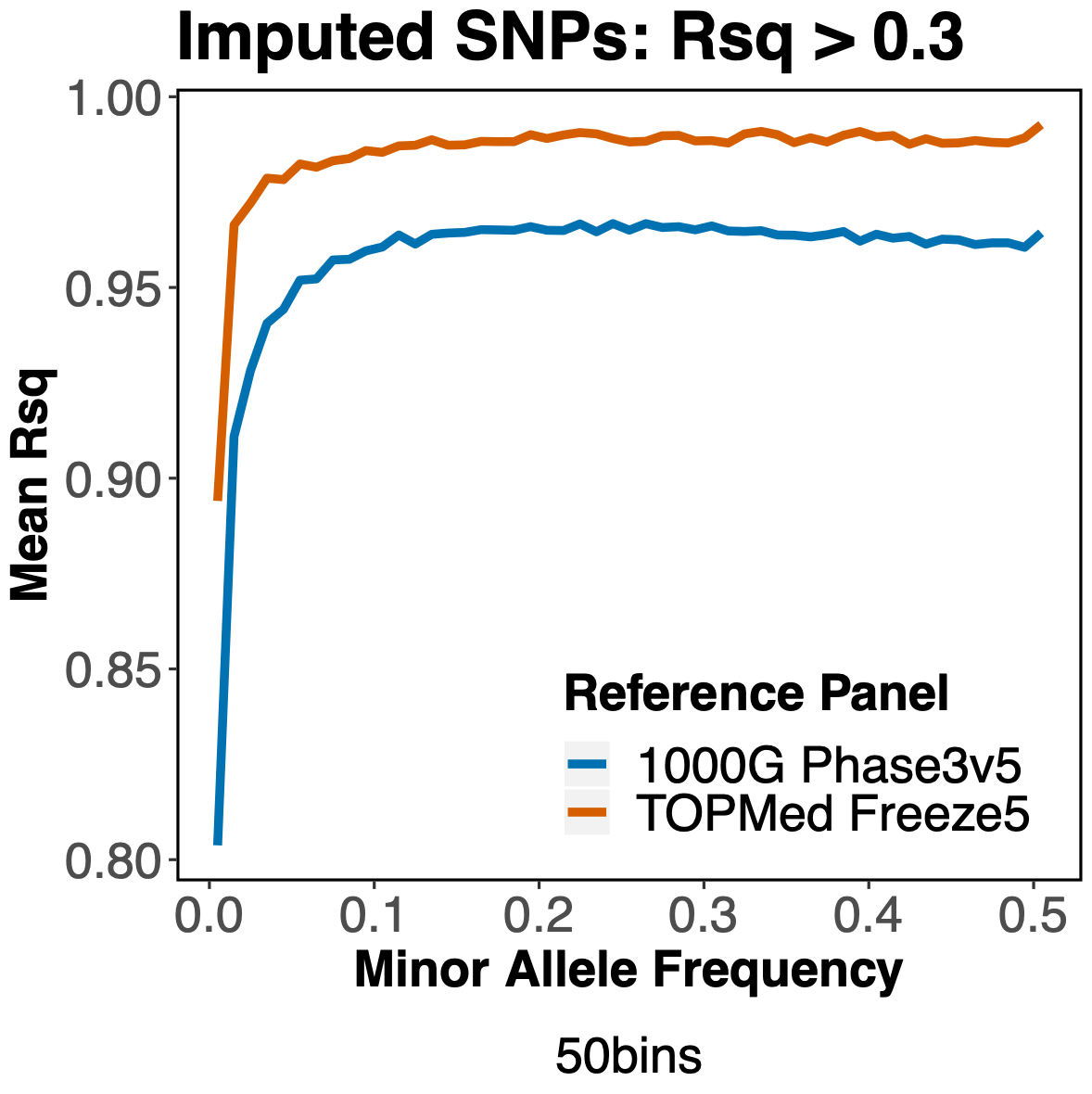
Since the TOPMed reference panel contains many more haplotypes than the 1000 Genomes panel, it appears we are getting better imputation of SNP with very low MAF.
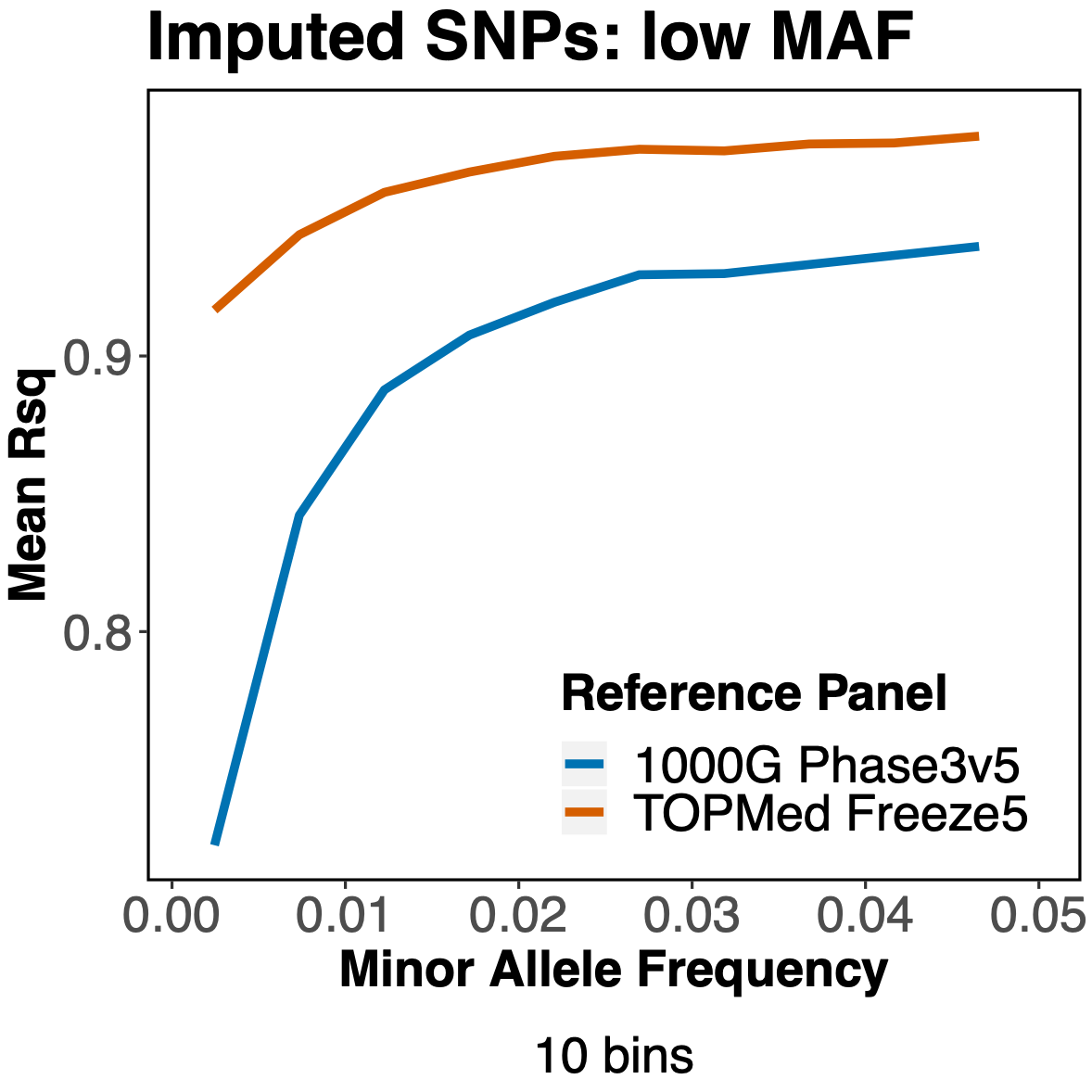
With these encouraging results, we decided to continue the analysis and apply other commonly used variant filters before doing our genetic association tests. Here is where we saw something unexpected. When filtering out variants with an MAF<0.01, TOPMed no longer yielded more imputed variants than 1000 Genomes. The final filter, variants with a sufficiently high Hardy-Weinberg equilibrium p-value (HWE > 1e-6), also showed more variants from the 1000 Genomes imputation.
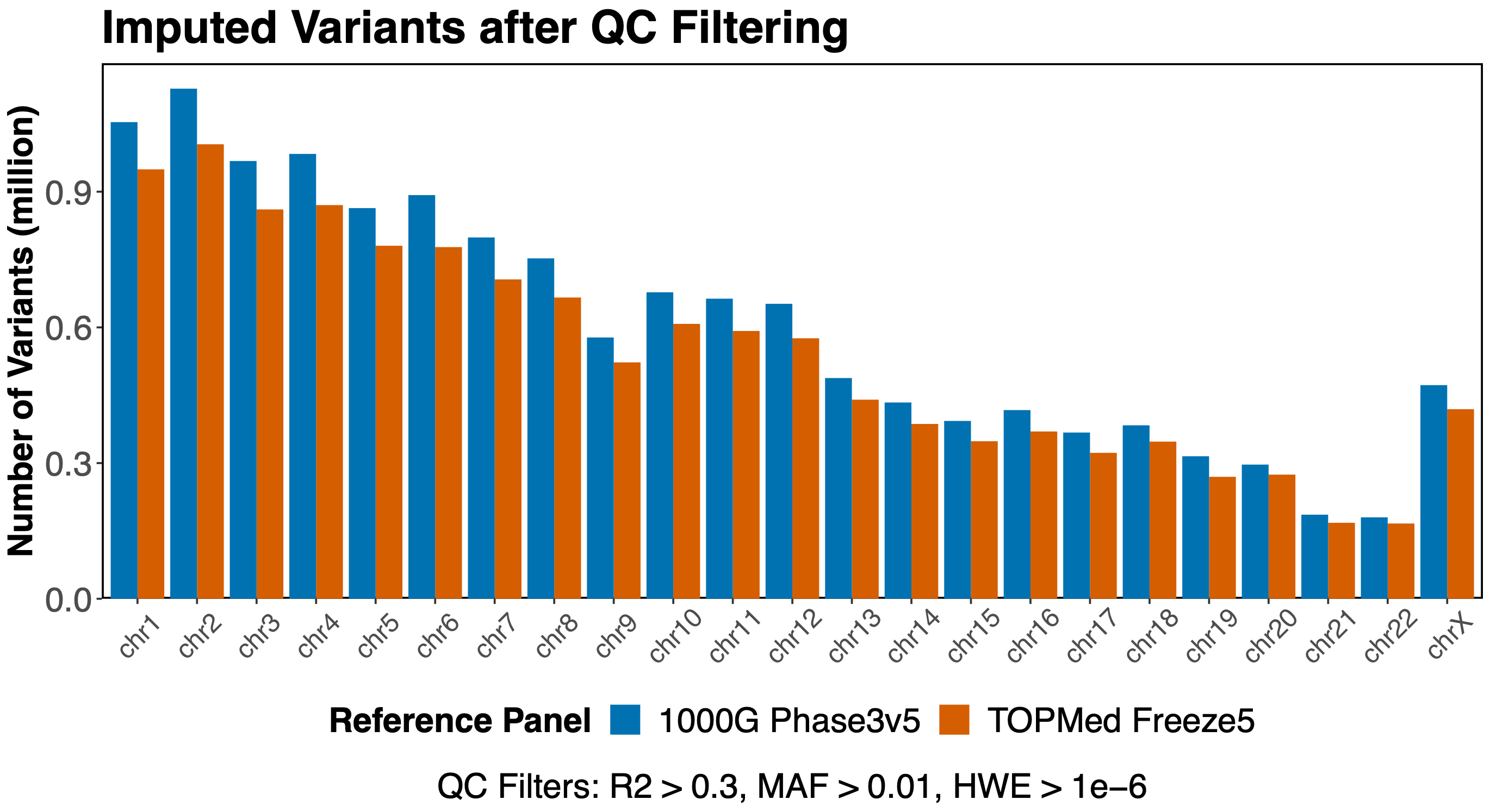
This effect is seen across the genome, the same for variants on any chromosome.
How could this be? It is possible that the TOPMed reference panel has enough genetic diversity to define more haplotypes at any give sequence in the genome. In this toy example, 1000 Genomes may have two haplotypes for the same genomic location that TOPMed defines three haplotypes. Some of the samples that were imputed to the 1000 Genomes rare haplotype may be imputed with greater accuracy to even rarer haplotypes in the TOPMed reference panel..

In this example, the 1000 Genomes imputed A/C variant has a MAF of 0.0184, which passes the variant filter cutoff of 0.01. The TOPMed imputed variants A/C and nearby A/G both have an MAF of 0.0092, which both fail the MAF cutoff. Indeed, when we look at the TOPMed imputed variants that are not passing the MAF cutoff, they have a lower MAF than the same variants imputed with 1000 Genomes.
But is this bad? As a data scientist, I hate to throw away data. I want to test more variants for genetic association. Indeed, that was the hope when we sought to use TOPMed instead of 1000 Genomes; we wanted to get more imputed variants by leveraging the greater genetic diversity in the TOPMed haplotypes. In the end, however, we tested fewer variants for genomic association. But this is rigorous science! The imputed variants we used in later association studies were imputed with greater confidence and contained fewer false positives. This in turn leads to fewer false positive genetic associations and a more robust data analysis.